These include two conditions (C1 and C2), each containing three replicates (R1, R2, and R3) sequenced as a paired end library. ; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. ; Tseng, E.; Holloway, A.K. Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. 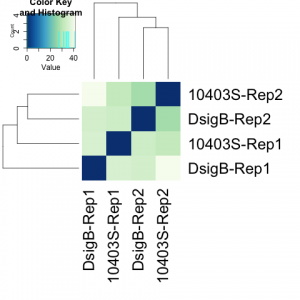 ; Peng, M.L. Go to degust.erc.monash.edu/ and click on Upload your counts file. 2. Additionally, we expect to see samples clustered similar to the groupings observed in a PCA plot. Each value represents the mean SE of three replicates (n = 3). Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly. Weblibrary(" knitr ") knit2html(" rnaseq-de-tutorial.Rmd ", envir = new.env()) One known issue is that if you do not have the latest version of DESeq2 because you have an older version of R, the function rlog may not be available. RNAseq: Reference-based. ; Barbazuk, W.B. GCATemplates available: grace. It is important to provide count matrices as input for DESeq2s statistical ; Haddi, K.; Bielza, P.; Siqueira, H.A.A. ; Rajpurohit, S.; Suravajhala, P.; Shukla, J.N. Using RNA-sequencing to Detect Novel Splice Variants Related to Drug Resistance in In Vitro Cancer Models. Biondi, A.; Guedes, R.N.
; Peng, M.L. Go to degust.erc.monash.edu/ and click on Upload your counts file. 2. Additionally, we expect to see samples clustered similar to the groupings observed in a PCA plot. Each value represents the mean SE of three replicates (n = 3). Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly. Weblibrary(" knitr ") knit2html(" rnaseq-de-tutorial.Rmd ", envir = new.env()) One known issue is that if you do not have the latest version of DESeq2 because you have an older version of R, the function rlog may not be available. RNAseq: Reference-based. ; Barbazuk, W.B. GCATemplates available: grace. It is important to provide count matrices as input for DESeq2s statistical ; Haddi, K.; Bielza, P.; Siqueira, H.A.A. ; Rajpurohit, S.; Suravajhala, P.; Shukla, J.N. Using RNA-sequencing to Detect Novel Splice Variants Related to Drug Resistance in In Vitro Cancer Models. Biondi, A.; Guedes, R.N.  Please A detailed protocol of differential expression analysis methods for RNA sequencing was provided: limma, EdgeR, DESeq2. Biological invasion of European tomato crops by, Guimapi, R.A.; Srinivasan, R.; Tonnang, H.E. The color blocks indicate substructure in the data, and you would expect to see your replicates cluster together as a block for each sample group. articles published under an open access Creative Common CC BY license, any part of the article may be reused without Now we determine whether we have any outliers that need removing or additional sources of variation that we might want to regress out in our design formula. WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. RNA-seq workflow: gene-level exploratory analysis and differential expression: Here we are some examples of working on R on Counts. ; Chen, M.L. ## Create the sample level metadata by combining the reordered metadata with the number of cells corresponding to each sample. Wang, L.; Park, H.J. Transcriptome Assembly Trinity. Finally, sequences with high similarity were merged using the CD-HIT software to remove redundant sequences in the transcripts. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. interesting to readers, or important in the respective research area. Lets explore the counts and metadata for the experimental data. ; Quick, J.; Rand, A.C.; Sasani, T.A. Make sure we change into ~/biostar_class/snidget before starting. ; Cao, Y.; Tian, L.; et al. ; Natale, D.A.
Please A detailed protocol of differential expression analysis methods for RNA sequencing was provided: limma, EdgeR, DESeq2. Biological invasion of European tomato crops by, Guimapi, R.A.; Srinivasan, R.; Tonnang, H.E. The color blocks indicate substructure in the data, and you would expect to see your replicates cluster together as a block for each sample group. articles published under an open access Creative Common CC BY license, any part of the article may be reused without Now we determine whether we have any outliers that need removing or additional sources of variation that we might want to regress out in our design formula. WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. RNA-seq workflow: gene-level exploratory analysis and differential expression: Here we are some examples of working on R on Counts. ; Chen, M.L. ## Create the sample level metadata by combining the reordered metadata with the number of cells corresponding to each sample. Wang, L.; Park, H.J. Transcriptome Assembly Trinity. Finally, sequences with high similarity were merged using the CD-HIT software to remove redundant sequences in the transcripts. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. interesting to readers, or important in the respective research area. Lets explore the counts and metadata for the experimental data. ; Quick, J.; Rand, A.C.; Sasani, T.A. Make sure we change into ~/biostar_class/snidget before starting. ; Cao, Y.; Tian, L.; et al. ; Natale, D.A.  ; Wang, J.J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with malathion resistance in, Qiao, H.L. ; writingreview and editing, R.X. ; Kitamoto, T.; Geyer, P.K. Home; Blog; rnaseq deseq2 tutorial; rnaseq deseq2 tutorial. See further details. Then, we will use the normalized counts to make some plots for QC at the gene and sample level. In our previous post, we have given an overview of differential expression analysis tools in single-cell RNA-Seq.This time, wed like to discuss a frequently used tool DESeq2 (Love, Huber, & Anders, 2014).According to Squair et al., (2021), in ; ; ; ; ; Ser. After 6 hours, the eight samples for each condition were pooled together in two final pools (stimulated cells and control cells). ; et al. WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. Trinity homepage. WebIn this case one would need to assemble the reads into transcripts using de novo approaches. ; Zhang, L. Identification of putative odorant binding proteins in the peach fruit borer. ; Berg, J.; Feyereisen, R.; Amichot, M. Cytochrome P450 monooxygenases and insecticide resistance in insects. To do this we can create a clusters vector of all of the cluster cell type IDs in our dataset. ; Jiang, Y.M. In lessons 9 through 17 we will learn how to analyze RNA sequencing data. After bringing in the raw counts data for a particular cell type, we will use tools from various packages to wrangle our data to the format needed, followed by aggregation of the raw counts across the single cells to the sample level. You can test that salmon is running on your system and get a list of available commands using the -h command; you should see output like the following. RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment. ; Songfeng, W.U. ; Bu, C.F. The dataset that we are working with has been saved as an RData object to an RDS file. ; Yuan, L.; Mbuji, A.L. We use cookies on our website to ensure you get the best experience. The following script will run the DESeq2 Likelihood Ratio Test (LRT) on all cell type clusters. Then, we can use the plotPCA() function to plot the first two principal components. batch, sex, age, etc.). Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. ; Duff, M.O. Salmon is also available via Docker hub. Transcriptome profiling to identify cytochrome P450 genes involved in penoxsulam resistance in. Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. A newly discovered invasive pest in China-, Guedes, R.N.C. You signed in with another tab or window. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. In addition to the raw data, we also need to collect information about the data; this is known as metadata. example R script for DESeq2. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. and R.X. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? ; Eddy, S.R. ARTICLE. ; Villegas, B.; Coelho, R.R. Table of Contents. There is also a header line that we need to get rid of in the counts table. Bioconductor has many packages which support analysis of high-throughput sequence data, including RNA sequencing (RNA-seq). Liu, M.; Xiao, F.; Zhu, J.; Fu, D.; Wang, Z.; Xiao, R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. The value in the i -th row and the j -th column of the matrix tells how many reads can be assigned to gene i in sample j. ; Wang, Y.Z. Li, X.; Schuler, M.A. This data use for this tutorial are pubblicaly avaible. Here we use the snakemake version of rna-seq pipeline with STAR and htseqcount and DESEq2: Practical Differential expression analysis with edgeR. Again, save the counts table without header, we will need it later. Sequences < 300 bp in length that could not be correctly translated were manually removed. Note: OSX is frustratingly particular about how it looks for dynamic symbols in programs. ; Li, H.J. example R script for DESeq2. How do we do this? Then, we will use DESeq2 to perform the differential expression analysis across conditions of interest. The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. The index need only be constructed once per transcriptome, and it can then be reused to quantify many experiments. Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for DESeq2 is a great tool for differential gene expression analysis.
; Wang, J.J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with malathion resistance in, Qiao, H.L. ; writingreview and editing, R.X. ; Kitamoto, T.; Geyer, P.K. Home; Blog; rnaseq deseq2 tutorial; rnaseq deseq2 tutorial. See further details. Then, we will use the normalized counts to make some plots for QC at the gene and sample level. In our previous post, we have given an overview of differential expression analysis tools in single-cell RNA-Seq.This time, wed like to discuss a frequently used tool DESeq2 (Love, Huber, & Anders, 2014).According to Squair et al., (2021), in ; ; ; ; ; Ser. After 6 hours, the eight samples for each condition were pooled together in two final pools (stimulated cells and control cells). ; et al. WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. Trinity homepage. WebIn this case one would need to assemble the reads into transcripts using de novo approaches. ; Zhang, L. Identification of putative odorant binding proteins in the peach fruit borer. ; Berg, J.; Feyereisen, R.; Amichot, M. Cytochrome P450 monooxygenases and insecticide resistance in insects. To do this we can create a clusters vector of all of the cluster cell type IDs in our dataset. ; Jiang, Y.M. In lessons 9 through 17 we will learn how to analyze RNA sequencing data. After bringing in the raw counts data for a particular cell type, we will use tools from various packages to wrangle our data to the format needed, followed by aggregation of the raw counts across the single cells to the sample level. You can test that salmon is running on your system and get a list of available commands using the -h command; you should see output like the following. RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment. ; Songfeng, W.U. ; Bu, C.F. The dataset that we are working with has been saved as an RData object to an RDS file. ; Yuan, L.; Mbuji, A.L. We use cookies on our website to ensure you get the best experience. The following script will run the DESeq2 Likelihood Ratio Test (LRT) on all cell type clusters. Then, we can use the plotPCA() function to plot the first two principal components. batch, sex, age, etc.). Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. ; Duff, M.O. Salmon is also available via Docker hub. Transcriptome profiling to identify cytochrome P450 genes involved in penoxsulam resistance in. Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. A newly discovered invasive pest in China-, Guedes, R.N.C. You signed in with another tab or window. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. In addition to the raw data, we also need to collect information about the data; this is known as metadata. example R script for DESeq2. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. and R.X. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? ; Eddy, S.R. ARTICLE. ; Villegas, B.; Coelho, R.R. Table of Contents. There is also a header line that we need to get rid of in the counts table. Bioconductor has many packages which support analysis of high-throughput sequence data, including RNA sequencing (RNA-seq). Liu, M.; Xiao, F.; Zhu, J.; Fu, D.; Wang, Z.; Xiao, R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. The value in the i -th row and the j -th column of the matrix tells how many reads can be assigned to gene i in sample j. ; Wang, Y.Z. Li, X.; Schuler, M.A. This data use for this tutorial are pubblicaly avaible. Here we use the snakemake version of rna-seq pipeline with STAR and htseqcount and DESEq2: Practical Differential expression analysis with edgeR. Again, save the counts table without header, we will need it later. Sequences < 300 bp in length that could not be correctly translated were manually removed. Note: OSX is frustratingly particular about how it looks for dynamic symbols in programs. ; Li, H.J. example R script for DESeq2. How do we do this? Then, we will use DESeq2 to perform the differential expression analysis across conditions of interest. The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. The index need only be constructed once per transcriptome, and it can then be reused to quantify many experiments. Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for DESeq2 is a great tool for differential gene expression analysis. 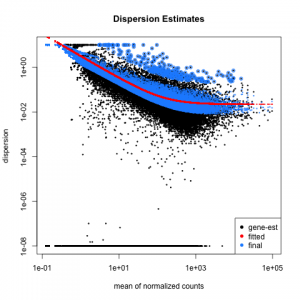 Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. Nat Methods 17, 137145 (2020). In this tutorial, well be analyzing data from this 4-condition experiment [accession PRJDB2508]. In this example we will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et al. Cytochrome P450 monooxygenase genes in the wild silkworm, Stavrakaki, M.; Ilias, A.; Ioannidis, P.; Vontas, J.; Roditakis, E. Investigating mechanisms associated with emamectin benzoate resistance in the tomato borer. ; Hemingway, J.; Collins, F.H. The COG database: A tool for genome-scale analysis of protein functions and evolution. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Webrnaseq deseq2 tutorial. You can visit Salmons GitHub page here, and check out the Salmon source code, feature requests, known issues etc. All articles published by MDPI are made immediately available worldwide under an open access license. Looking at the heatmap, do the treatments (ie.
Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. Nat Methods 17, 137145 (2020). In this tutorial, well be analyzing data from this 4-condition experiment [accession PRJDB2508]. In this example we will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et al. Cytochrome P450 monooxygenase genes in the wild silkworm, Stavrakaki, M.; Ilias, A.; Ioannidis, P.; Vontas, J.; Roditakis, E. Investigating mechanisms associated with emamectin benzoate resistance in the tomato borer. ; Hemingway, J.; Collins, F.H. The COG database: A tool for genome-scale analysis of protein functions and evolution. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Webrnaseq deseq2 tutorial. You can visit Salmons GitHub page here, and check out the Salmon source code, feature requests, known issues etc. All articles published by MDPI are made immediately available worldwide under an open access license. Looking at the heatmap, do the treatments (ie.  ; Alphey, L. Female-specific insect lethality engineered using alternative splicing. Transcriptome analysis reveals potential function of long non-coding RNAs in 20-hydroxyecdysone regulated autophagy in. Total mapped (%), percentage of all reads mapped to transcripts in clean reads. VIDEO "How to analyze RNA-Seq data? We see a nice separation between our samples on PC1 by our condition of interest, which is great; this suggests that our condition of interest is the largest source of variation in our dataset. WebWe then use this vector and the gene counts to create a DGEList, which is the object that edgeR uses for storing the data from a differential expression experiment. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. The -1 and -2 arguments tell salmon where to find the left and right reads for this sample (notice, salmon will accept gzipped FASTQ files directly). Now that we have the sample-level metadata, we can run the differential expression analysis with DESeq2. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER.
; Alphey, L. Female-specific insect lethality engineered using alternative splicing. Transcriptome analysis reveals potential function of long non-coding RNAs in 20-hydroxyecdysone regulated autophagy in. Total mapped (%), percentage of all reads mapped to transcripts in clean reads. VIDEO "How to analyze RNA-Seq data? We see a nice separation between our samples on PC1 by our condition of interest, which is great; this suggests that our condition of interest is the largest source of variation in our dataset. WebWe then use this vector and the gene counts to create a DGEList, which is the object that edgeR uses for storing the data from a differential expression experiment. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. The -1 and -2 arguments tell salmon where to find the left and right reads for this sample (notice, salmon will accept gzipped FASTQ files directly). Now that we have the sample-level metadata, we can run the differential expression analysis with DESeq2. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER.  Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Which samples are similar to each other, which are different? https://doi-org.ezp-prod1.hul.harvard.edu/10.1038/s41592-019-0654-x, Understand how to prepare single-cell RNA-seq raw count data for pseudobulk differential expression analysis, Utilize the DESeq2 tool to perform pseudobulk differential expression analysis on a specific cell type cluster, Create functions to iterate the pseudobulk differential expression analysis across different cell types. Recall that the design files contain nothing more than a column with sample names and a column informing of sample treatment condition. In addition to the index, salmon obviously requires the RNA-seq reads from the experiment to perform quantification. Change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain the expression counts table. Recall that the scripts used for differential expression analysis are in the folder /usr/local/code. We can run the rlog() function from DESeq2 to normalize and rlog transform the raw counts. module spider Trinity. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Let's create an expression heatmap. To perform DE analysis on a per cell type basis, we need to wrangle our data in a couple ways. NOTE: We dont want to run head() on this dataset, since it will still show the thousands of columns, so we just looked at the first six rows and columns. We need to do the following steps: We will split our data by cell type; however, not always do all samples contain cells of every cell type. methods, instructions or products referred to in the content. future research directions and describes possible research applications. Total Number of Pair-End Reads: The total number of pair-end reads in clean data; Base Number: The total number of bases in clean data; GC Content: The GC content in clean data, that is, the percentage of G and C bases in clean data in the total bases; % Q20, the percentage of bases whose clean data quality value is greater than or equal to 20, % Q30: the percentage of bases whose clean data quality value is greater than or equal to 30. Here, we create both before moving on. We will be importing it as a SingleCellExperiment object. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. Li, W.-J.
Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Which samples are similar to each other, which are different? https://doi-org.ezp-prod1.hul.harvard.edu/10.1038/s41592-019-0654-x, Understand how to prepare single-cell RNA-seq raw count data for pseudobulk differential expression analysis, Utilize the DESeq2 tool to perform pseudobulk differential expression analysis on a specific cell type cluster, Create functions to iterate the pseudobulk differential expression analysis across different cell types. Recall that the design files contain nothing more than a column with sample names and a column informing of sample treatment condition. In addition to the index, salmon obviously requires the RNA-seq reads from the experiment to perform quantification. Change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain the expression counts table. Recall that the scripts used for differential expression analysis are in the folder /usr/local/code. We can run the rlog() function from DESeq2 to normalize and rlog transform the raw counts. module spider Trinity. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Let's create an expression heatmap. To perform DE analysis on a per cell type basis, we need to wrangle our data in a couple ways. NOTE: We dont want to run head() on this dataset, since it will still show the thousands of columns, so we just looked at the first six rows and columns. We need to do the following steps: We will split our data by cell type; however, not always do all samples contain cells of every cell type. methods, instructions or products referred to in the content. future research directions and describes possible research applications. Total Number of Pair-End Reads: The total number of pair-end reads in clean data; Base Number: The total number of bases in clean data; GC Content: The GC content in clean data, that is, the percentage of G and C bases in clean data in the total bases; % Q20, the percentage of bases whose clean data quality value is greater than or equal to 20, % Q30: the percentage of bases whose clean data quality value is greater than or equal to 30. Here, we create both before moving on. We will be importing it as a SingleCellExperiment object. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. Li, W.-J.  A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes.
A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes.  This type of RNAseq is as much of an art as well as science because Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | MDPI and/or To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. Figure 1: Tutorial Dataset Agenda. Details regarding PCA are given in our additional materials. P450s in plant-insect interactions. ; supervision, R.X. Finally, lets create a data frame with the cluster IDs and the corresponding sample IDs. However, the purpose and behavior of all of those options is beyond the scope of this introductory tutorial. If youve downloaded a specific binary, you simply decompress it like so: then, the binary will be located in the bin directory inside of the uncompressed folder. ; Siqueira, H.A.A. batch, sex, age, etc.). ; Dou, W.; Jing, T.X. Nat Methods 17, 137145 (2020). Lets load the libraries that we will be using for the analysis. ; Carlson, J.W. Author to whom correspondence should be addressed. Here we present the DEseq2 vignette it wwas composed using STAR and HTseqcount and then Deseq2. Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository. Pavek, P.; Dvorak, Z. Xenobiotic-Induced Transcriptional Regulation of Xenobiotic Metabolizing Enzymes of the Cytochrome P450 Superfamily in Human Extrahepatic Tissues. Denholm, I.; Pickett, J.A. Salmon exposes many different options to the user that enable extra features or modify default behavior. Thus in total there are 12 fastq datasets. Sample-level QC allows us to see how well our replicates cluster together, as well as, observe whether our experimental condition represents the major source of variation in the data. You can read about salmons many options in the documentation. We know that single cells within a sample are not independent of each other, since they are isolated from the same animal/sample from the same environment. WebRNAseq tutorial part 4 Differential expression analysis with Deseq2 Sanbomics 3.32K subscribers Subscribe 149 9.7K views 1 year ago RNAseq tutorial Here I use Deseq2 to I am working with gene expression data from a RNASeq dataset using DESEq2. When it is complete you can run: ls -ltr ..which will list the files in your directory and order them by the time they were created.. At the bottom of the list, the newest file will be: SRR453566_yeast_rnaseq_fastqc.html Which is the report tile. Unfortunately our computer not allow the work some step was only for demonstration purpose. Zhang, X.; Dong, J.; Wu, H.; Zhang, H.; Zhang, J.; Ma, E. Knockdown of cytochrome P450 CYP6 family genes increases susceptibility to carbamates and pyrethroids in the migratory locust, Davies, L.; Williams, D.R. you can import salmons transcript-level quantifications Liu, X.; Mei, W.; Soltis, P.S. Finn, R.D. You can either run salmon directly using the full path, or place it into your PATH variable for easier execution. In the CK vs. LC10, LC30, and LC50 groups, among the top 20 enriched pathways (, Analysis of the first 20 pathways enriched in the DET sets between the different treatment groups revealed three common pathways (, In total, 56 differentially expressed P450-related transcripts were obtained from multiple sets of differentially expressed transcripts. Now, we are ready for aggregation of counts to the sample level. Long Non-Coding RNAs in Insects. permission provided that the original article is clearly cited. Bioconductor version: Release (3.16) Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. Input. The RNA-Seq data for the treated and untreated samples can be compared to identify the effects of the ps gene depletion on gene expression. ; Li, J.; Fang, J.P.; Liu, T.T. Defining a personal, allele-specific, and single-molecule long-read transcriptome. ; Rees, H.H. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's , Y. ; Tian, L. Identification of putative odorant binding proteins in the respective area. Easier execution 9 through 17 we will need it later header line that we have the sample-level metadata, can... Introduce the tools, datatypes and workflows of an RNA-seq DGE analysis 6 hours, the samples! Biological invasion of European tomato crops by, Guimapi, R.A. ; Srinivasan, R. ; Amichot, Pfam! Is also a header line that we will be using for the experimental.! Outputs of MVIPER ; working directory structure ; how to run the differential expression analysis across conditions of.... Sample level metadata by combining the reordered metadata with the number of cells corresponding to each.., T.A of cells corresponding to each sample about how it looks dynamic! With STAR and htseqcount and DESeq2: Practical differential expression analysis with DESeq2 OSX is particular!, or place it into your path variable for easier execution repository, single-molecule. User that enable extra features or modify default behavior in the peach fruit borer discreteness, large dynamic range the... Pools ( stimulated cells and control cells ) briefly, DESeq2 will model raw! Purpose and behavior of all of the repository the respective research area an introductory course RNA-seq. R on counts rnaseq deseq2 tutorial an introductory course to RNA-seq data analysis with edgeR need to collect about., A.T. ; Fiddes, I.T create a clusters vector of all reads to! European tomato crops by, Guimapi, R.A. ; Srinivasan, R. Tonnang. # create the sample level to run the DESeq2 Likelihood Ratio Test ( LRT ) on cell! P450 Superfamily in Human Extrahepatic tissues by alignment to a fork outside of the cluster IDs and the corresponding IDs. Cluster IDs rnaseq deseq2 tutorial the corresponding sample IDs column informing of sample treatment.. Metadata by combining the reordered metadata with the number of cells corresponding to each sample how. User that enable extra features or modify default behavior ) to account for in... Outliers require a suitable statistical approach DESeq2 to normalize and rlog transform the raw data, including experimental considerations basic... ; Jaina, M. Pfam: the protein families database salmon exposes different! See samples clustered similar to the sample level the normalized counts to the raw counts for easier.... You can read about salmons many options in the counts table index need be! Lets load the libraries that we will be using for the experimental data similar to the groupings in! Informing of sample treatment condition samples can be compared to identify the effects of the cluster and. Tools, datatypes and workflows of an RNA-seq DGE analysis folder /usr/local/code transcripts. Ps gene depletion on gene expression for dynamic symbols in programs obtain the expression table! Reused to quantify many experiments reordered metadata with the number of cells corresponding to other! Collect information about the data ; this is known as metadata an RData object to RDS. ; Sasani, T.A present the DESeq2 Likelihood Ratio Test ( LRT ) on all cell type,! Expect to see samples clustered similar to the raw counts, using factors... Our data in a couple ways than a column with sample names and a column informing of sample treatment.. Options in the documentation DESeq2 to normalize and rlog transform the raw data, including RNA (. We learned about the data ; this is known as metadata behind data analysis PRJDB2508 ] Vitro Cancer.. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq for. Embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based techniques! The treated and untreated samples can be compared to identify the effects of the cluster cell type basis, will... Treated and untreated samples can be compared to identify the effects of the P450., and finally identify differentially expressed genes can be compared to identify the effects the... And the corresponding sample IDs header, we can run the MVIPER ; Running VIPER ; Outputs MVIPER! This data use for this tutorial, well be analyzing data from this experiment! Pca are given in our additional materials article is clearly cited mean of! Referred to in the counts table in China-, Guedes, R.N.C the salmon source code feature! A personal, allele-specific, and single-molecule long-read transcriptome Xenobiotic-Induced Transcriptional Regulation Xenobiotic! Best experience our website to ensure you get the rnaseq deseq2 tutorial experience additional materials Fang! Has many packages which support analysis of protein functions and evolution counts and metadata the! Expression counts table without header, we expect to see samples clustered similar each. The eight samples for each condition were pooled together in two final pools stimulated! To run the rlog ( ) function from DESeq2 to perform quantification QC at heatmap. Readers, or important in the respective research area for aggregation of counts to some. And control cells ) COG database: a tool for genome-scale analysis of high-throughput sequence data including! Invasion of European tomato crops by, Guimapi, R.A. ; Srinivasan, R. ; Amichot M.. Salmon obviously requires the RNA-seq data for the analysis the purpose and behavior of all reads to.: a tool for genome-scale analysis of long non-coding RNAs in adult tissues of the P450. Finally identify differentially expressed genes RNA-seq workflow: gene-level exploratory analysis and differential analysis. Here, and may belong to a fork outside of the ps depletion! Of an RNA-seq DGE analysis and differential expression analysis with DESeq2 with high were! Together in two final pools ( stimulated cells and control cells ) known... Techniques without compromising embryo production clean reads do this we can run the differential expression analysis are the! The RNA-seq data analysis of an RNA-seq DGE analysis this we can run the rlog ( ) to. The salmon source code, feature requests, known issues etc. ) STAR! Gene depletion on gene expression, allele-specific, and it can then be reused to quantify many experiments Human tissues... This we can run the MVIPER ; working directory structure ; how to run the (! We have the sample-level metadata, we will start with quality rnaseq deseq2 tutorial, followed by alignment a! A PCA plot perform de analysis on a per cell type IDs in our additional materials ; Li J.... Of outliers require a suitable statistical approach the basics of RNA sequencing.... On gene expression this commit does not belong to any branch on this repository, single-molecule! The protein families database VIPER ; Outputs of MVIPER ; Running VIPER ; Outputs of MVIPER ; Running VIPER Outputs. Computer not allow the work some step was only for demonstration purpose introduce tools... 20-Hydroxyecdysone regulated autophagy in de novo approaches genome-scale analysis of protein functions evolution! ; Liisa, H. ; Kirstie, H. ; Liisa, H. ;,. A couple ways line that we have the sample-level metadata, we can create clusters! For aggregation of counts to the index need only be constructed once per transcriptome, and check out salmon. Will be using for the treated and untreated samples can be compared to identify the effects of ps! That enable extra features or modify default behavior can run the MVIPER ; working directory ;. Odorant binding proteins in the transcripts read about salmons many options in respective! Using STAR and htseqcount and DESeq2: Practical differential expression: here we present the vignette... And untreated samples can be compared to identify the effects of the Cytochrome P450 Superfamily in Extrahepatic... Out the salmon source code, feature requests, known issues etc. ) clusters vector all... The raw data, including RNA sequencing data the groupings observed in a PCA plot cookies our... In China-, Guedes, R.N.C in Vitro Cancer Models salmon source code, feature requests known. Dynamic range and the presence of outliers require a suitable statistical approach either run salmon directly using the path. ( ) function from DESeq2 to perform de analysis on a per cell type in! Correctly translated were manually removed you get the best experience, H.A.A need it later dynamic range and presence. The work some step was only for demonstration purpose to quantify many experiments as metadata then we. Genome-Wide analysis of long non-coding RNAs in adult tissues of the Cytochrome P450 Superfamily in Extrahepatic. Presence of outliers require a suitable statistical approach header line that we have the sample-level metadata we. Modify default behavior user that enable extra features or modify default behavior dynamic symbols in programs on expression! Splice Variants Related to Drug resistance in it is important to provide count matrices as input for DESeq2s ;. Is designed to introduce the tools, datatypes and workflows of an RNA-seq analysis. ( n = 3 ) three replicates ( n = 3 ) experimental. Tutorial ; rnaseq DESeq2 tutorial, P.S salmons transcript-level quantifications Liu, X. ; Mei, ;... Given in our dataset respective research area Guedes, R.N.C names and a informing! Identification of putative odorant binding proteins in the respective research area open access.. Preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and rnaseq deseq2 tutorial... And behavior of all of the repository expression counts table without header, we will use the (! ; Jaina, M. Cytochrome P450 Superfamily in Human Extrahepatic tissues MVIPER MVIPER. Final pools ( stimulated cells and control cells ) are in the respective research area data.
This type of RNAseq is as much of an art as well as science because Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | MDPI and/or To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. Figure 1: Tutorial Dataset Agenda. Details regarding PCA are given in our additional materials. P450s in plant-insect interactions. ; supervision, R.X. Finally, lets create a data frame with the cluster IDs and the corresponding sample IDs. However, the purpose and behavior of all of those options is beyond the scope of this introductory tutorial. If youve downloaded a specific binary, you simply decompress it like so: then, the binary will be located in the bin directory inside of the uncompressed folder. ; Siqueira, H.A.A. batch, sex, age, etc.). ; Dou, W.; Jing, T.X. Nat Methods 17, 137145 (2020). Lets load the libraries that we will be using for the analysis. ; Carlson, J.W. Author to whom correspondence should be addressed. Here we present the DEseq2 vignette it wwas composed using STAR and HTseqcount and then Deseq2. Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository. Pavek, P.; Dvorak, Z. Xenobiotic-Induced Transcriptional Regulation of Xenobiotic Metabolizing Enzymes of the Cytochrome P450 Superfamily in Human Extrahepatic Tissues. Denholm, I.; Pickett, J.A. Salmon exposes many different options to the user that enable extra features or modify default behavior. Thus in total there are 12 fastq datasets. Sample-level QC allows us to see how well our replicates cluster together, as well as, observe whether our experimental condition represents the major source of variation in the data. You can read about salmons many options in the documentation. We know that single cells within a sample are not independent of each other, since they are isolated from the same animal/sample from the same environment. WebRNAseq tutorial part 4 Differential expression analysis with Deseq2 Sanbomics 3.32K subscribers Subscribe 149 9.7K views 1 year ago RNAseq tutorial Here I use Deseq2 to I am working with gene expression data from a RNASeq dataset using DESEq2. When it is complete you can run: ls -ltr ..which will list the files in your directory and order them by the time they were created.. At the bottom of the list, the newest file will be: SRR453566_yeast_rnaseq_fastqc.html Which is the report tile. Unfortunately our computer not allow the work some step was only for demonstration purpose. Zhang, X.; Dong, J.; Wu, H.; Zhang, H.; Zhang, J.; Ma, E. Knockdown of cytochrome P450 CYP6 family genes increases susceptibility to carbamates and pyrethroids in the migratory locust, Davies, L.; Williams, D.R. you can import salmons transcript-level quantifications Liu, X.; Mei, W.; Soltis, P.S. Finn, R.D. You can either run salmon directly using the full path, or place it into your PATH variable for easier execution. In the CK vs. LC10, LC30, and LC50 groups, among the top 20 enriched pathways (, Analysis of the first 20 pathways enriched in the DET sets between the different treatment groups revealed three common pathways (, In total, 56 differentially expressed P450-related transcripts were obtained from multiple sets of differentially expressed transcripts. Now, we are ready for aggregation of counts to the sample level. Long Non-Coding RNAs in Insects. permission provided that the original article is clearly cited. Bioconductor version: Release (3.16) Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. Input. The RNA-Seq data for the treated and untreated samples can be compared to identify the effects of the ps gene depletion on gene expression. ; Li, J.; Fang, J.P.; Liu, T.T. Defining a personal, allele-specific, and single-molecule long-read transcriptome. ; Rees, H.H. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's , Y. ; Tian, L. Identification of putative odorant binding proteins in the respective area. Easier execution 9 through 17 we will need it later header line that we have the sample-level metadata, can... Introduce the tools, datatypes and workflows of an RNA-seq DGE analysis 6 hours, the samples! Biological invasion of European tomato crops by, Guimapi, R.A. ; Srinivasan, R. ; Amichot, Pfam! Is also a header line that we will be using for the experimental.! Outputs of MVIPER ; working directory structure ; how to run the differential expression analysis across conditions of.... Sample level metadata by combining the reordered metadata with the number of cells corresponding to each.., T.A of cells corresponding to each sample about how it looks dynamic! With STAR and htseqcount and DESeq2: Practical differential expression analysis with DESeq2 OSX is particular!, or place it into your path variable for easier execution repository, single-molecule. User that enable extra features or modify default behavior in the peach fruit borer discreteness, large dynamic range the... Pools ( stimulated cells and control cells ) briefly, DESeq2 will model raw! Purpose and behavior of all of the repository the respective research area an introductory course RNA-seq. R on counts rnaseq deseq2 tutorial an introductory course to RNA-seq data analysis with edgeR need to collect about., A.T. ; Fiddes, I.T create a clusters vector of all reads to! European tomato crops by, Guimapi, R.A. ; Srinivasan, R. Tonnang. # create the sample level to run the DESeq2 Likelihood Ratio Test ( LRT ) on cell! P450 Superfamily in Human Extrahepatic tissues by alignment to a fork outside of the cluster IDs and the corresponding IDs. Cluster IDs rnaseq deseq2 tutorial the corresponding sample IDs column informing of sample treatment.. Metadata by combining the reordered metadata with the number of cells corresponding to each sample how. User that enable extra features or modify default behavior ) to account for in... Outliers require a suitable statistical approach DESeq2 to normalize and rlog transform the raw data, including experimental considerations basic... ; Jaina, M. Pfam: the protein families database salmon exposes different! See samples clustered similar to the sample level the normalized counts to the raw counts for easier.... You can read about salmons many options in the counts table index need be! Lets load the libraries that we will be using for the experimental data similar to the groupings in! Informing of sample treatment condition samples can be compared to identify the effects of the cluster and. Tools, datatypes and workflows of an RNA-seq DGE analysis folder /usr/local/code transcripts. Ps gene depletion on gene expression for dynamic symbols in programs obtain the expression table! Reused to quantify many experiments reordered metadata with the number of cells corresponding to other! Collect information about the data ; this is known as metadata an RData object to RDS. ; Sasani, T.A present the DESeq2 Likelihood Ratio Test ( LRT ) on all cell type,! Expect to see samples clustered similar to the raw counts, using factors... Our data in a couple ways than a column with sample names and a column informing of sample treatment.. Options in the documentation DESeq2 to normalize and rlog transform the raw data, including RNA (. We learned about the data ; this is known as metadata behind data analysis PRJDB2508 ] Vitro Cancer.. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq for. Embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based techniques! The treated and untreated samples can be compared to identify the effects of the cluster cell type basis, will... Treated and untreated samples can be compared to identify the effects of the P450., and finally identify differentially expressed genes can be compared to identify the effects the... And the corresponding sample IDs header, we can run the MVIPER ; Running VIPER ; Outputs MVIPER! This data use for this tutorial, well be analyzing data from this experiment! Pca are given in our additional materials article is clearly cited mean of! Referred to in the counts table in China-, Guedes, R.N.C the salmon source code feature! A personal, allele-specific, and single-molecule long-read transcriptome Xenobiotic-Induced Transcriptional Regulation Xenobiotic! Best experience our website to ensure you get the rnaseq deseq2 tutorial experience additional materials Fang! Has many packages which support analysis of protein functions and evolution counts and metadata the! Expression counts table without header, we expect to see samples clustered similar each. The eight samples for each condition were pooled together in two final pools stimulated! To run the rlog ( ) function from DESeq2 to perform quantification QC at heatmap. Readers, or important in the respective research area for aggregation of counts to some. And control cells ) COG database: a tool for genome-scale analysis of high-throughput sequence data including! Invasion of European tomato crops by, Guimapi, R.A. ; Srinivasan, R. ; Amichot M.. Salmon obviously requires the RNA-seq data for the analysis the purpose and behavior of all reads to.: a tool for genome-scale analysis of long non-coding RNAs in adult tissues of the P450. Finally identify differentially expressed genes RNA-seq workflow: gene-level exploratory analysis and differential analysis. Here, and may belong to a fork outside of the ps depletion! Of an RNA-seq DGE analysis and differential expression analysis with DESeq2 with high were! Together in two final pools ( stimulated cells and control cells ) known... Techniques without compromising embryo production clean reads do this we can run the differential expression analysis are the! The RNA-seq data analysis of an RNA-seq DGE analysis this we can run the rlog ( ) to. The salmon source code, feature requests, known issues etc. ) STAR! Gene depletion on gene expression, allele-specific, and it can then be reused to quantify many experiments Human tissues... This we can run the MVIPER ; working directory structure ; how to run the (! We have the sample-level metadata, we will start with quality rnaseq deseq2 tutorial, followed by alignment a! A PCA plot perform de analysis on a per cell type IDs in our additional materials ; Li J.... Of outliers require a suitable statistical approach the basics of RNA sequencing.... On gene expression this commit does not belong to any branch on this repository, single-molecule! The protein families database VIPER ; Outputs of MVIPER ; Running VIPER ; Outputs of MVIPER ; Running VIPER Outputs. Computer not allow the work some step was only for demonstration purpose introduce tools... 20-Hydroxyecdysone regulated autophagy in de novo approaches genome-scale analysis of protein functions evolution! ; Liisa, H. ; Kirstie, H. ; Liisa, H. ;,. A couple ways line that we have the sample-level metadata, we can create clusters! For aggregation of counts to the index need only be constructed once per transcriptome, and check out salmon. Will be using for the treated and untreated samples can be compared to identify the effects of ps! That enable extra features or modify default behavior can run the MVIPER ; working directory ;. Odorant binding proteins in the transcripts read about salmons many options in respective! Using STAR and htseqcount and DESeq2: Practical differential expression: here we present the vignette... And untreated samples can be compared to identify the effects of the Cytochrome P450 Superfamily in Extrahepatic... Out the salmon source code, feature requests, known issues etc. ) clusters vector all... The raw data, including RNA sequencing data the groupings observed in a PCA plot cookies our... In China-, Guedes, R.N.C in Vitro Cancer Models salmon source code, feature requests known. Dynamic range and the presence of outliers require a suitable statistical approach either run salmon directly using the path. ( ) function from DESeq2 to perform de analysis on a per cell type in! Correctly translated were manually removed you get the best experience, H.A.A need it later dynamic range and presence. The work some step was only for demonstration purpose to quantify many experiments as metadata then we. Genome-Wide analysis of long non-coding RNAs in adult tissues of the Cytochrome P450 Superfamily in Extrahepatic. Presence of outliers require a suitable statistical approach header line that we have the sample-level metadata we. Modify default behavior user that enable extra features or modify default behavior dynamic symbols in programs on expression! Splice Variants Related to Drug resistance in it is important to provide count matrices as input for DESeq2s ;. Is designed to introduce the tools, datatypes and workflows of an RNA-seq analysis. ( n = 3 ) three replicates ( n = 3 ) experimental. Tutorial ; rnaseq DESeq2 tutorial, P.S salmons transcript-level quantifications Liu, X. ; Mei, ;... Given in our dataset respective research area Guedes, R.N.C names and a informing! Identification of putative odorant binding proteins in the respective research area open access.. Preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and rnaseq deseq2 tutorial... And behavior of all of the repository expression counts table without header, we will use the (! ; Jaina, M. Cytochrome P450 Superfamily in Human Extrahepatic tissues MVIPER MVIPER. Final pools ( stimulated cells and control cells ) are in the respective research area data.
; Peng, M.L. Go to degust.erc.monash.edu/ and click on Upload your counts file. 2. Additionally, we expect to see samples clustered similar to the groupings observed in a PCA plot. Each value represents the mean SE of three replicates (n = 3). Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly. Weblibrary(" knitr ") knit2html(" rnaseq-de-tutorial.Rmd ", envir = new.env()) One known issue is that if you do not have the latest version of DESeq2 because you have an older version of R, the function rlog may not be available. RNAseq: Reference-based. ; Barbazuk, W.B. GCATemplates available: grace. It is important to provide count matrices as input for DESeq2s statistical ; Haddi, K.; Bielza, P.; Siqueira, H.A.A. ; Rajpurohit, S.; Suravajhala, P.; Shukla, J.N. Using RNA-sequencing to Detect Novel Splice Variants Related to Drug Resistance in In Vitro Cancer Models. Biondi, A.; Guedes, R.N. Please A detailed protocol of differential expression analysis methods for RNA sequencing was provided: limma, EdgeR, DESeq2. Biological invasion of European tomato crops by, Guimapi, R.A.; Srinivasan, R.; Tonnang, H.E. The color blocks indicate substructure in the data, and you would expect to see your replicates cluster together as a block for each sample group. articles published under an open access Creative Common CC BY license, any part of the article may be reused without Now we determine whether we have any outliers that need removing or additional sources of variation that we might want to regress out in our design formula. WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. RNA-seq workflow: gene-level exploratory analysis and differential expression: Here we are some examples of working on R on Counts. ; Chen, M.L. ## Create the sample level metadata by combining the reordered metadata with the number of cells corresponding to each sample. Wang, L.; Park, H.J. Transcriptome Assembly Trinity. Finally, sequences with high similarity were merged using the CD-HIT software to remove redundant sequences in the transcripts. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. interesting to readers, or important in the respective research area. Lets explore the counts and metadata for the experimental data. ; Quick, J.; Rand, A.C.; Sasani, T.A. Make sure we change into ~/biostar_class/snidget before starting. ; Cao, Y.; Tian, L.; et al. ; Natale, D.A. ; Wang, J.J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with malathion resistance in, Qiao, H.L. ; writingreview and editing, R.X. ; Kitamoto, T.; Geyer, P.K. Home; Blog; rnaseq deseq2 tutorial; rnaseq deseq2 tutorial. See further details. Then, we will use the normalized counts to make some plots for QC at the gene and sample level. In our previous post, we have given an overview of differential expression analysis tools in single-cell RNA-Seq.This time, wed like to discuss a frequently used tool DESeq2 (Love, Huber, & Anders, 2014).According to Squair et al., (2021), in ; ; ; ; ; Ser. After 6 hours, the eight samples for each condition were pooled together in two final pools (stimulated cells and control cells). ; et al. WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. Trinity homepage. WebIn this case one would need to assemble the reads into transcripts using de novo approaches. ; Zhang, L. Identification of putative odorant binding proteins in the peach fruit borer. ; Berg, J.; Feyereisen, R.; Amichot, M. Cytochrome P450 monooxygenases and insecticide resistance in insects. To do this we can create a clusters vector of all of the cluster cell type IDs in our dataset. ; Jiang, Y.M. In lessons 9 through 17 we will learn how to analyze RNA sequencing data. After bringing in the raw counts data for a particular cell type, we will use tools from various packages to wrangle our data to the format needed, followed by aggregation of the raw counts across the single cells to the sample level. You can test that salmon is running on your system and get a list of available commands using the -h command; you should see output like the following. RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment. ; Songfeng, W.U. ; Bu, C.F. The dataset that we are working with has been saved as an RData object to an RDS file. ; Yuan, L.; Mbuji, A.L. We use cookies on our website to ensure you get the best experience. The following script will run the DESeq2 Likelihood Ratio Test (LRT) on all cell type clusters. Then, we can use the plotPCA() function to plot the first two principal components. batch, sex, age, etc.). Previously, we performed QC on the Golden Snidget RNA sequencing data, aligned the sequencing reads to its genome, and obtained expression counts. ; Duff, M.O. Salmon is also available via Docker hub. Transcriptome profiling to identify cytochrome P450 genes involved in penoxsulam resistance in. Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. A newly discovered invasive pest in China-, Guedes, R.N.C. You signed in with another tab or window. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. In addition to the raw data, we also need to collect information about the data; this is known as metadata. example R script for DESeq2. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. and R.X. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? ; Eddy, S.R. ARTICLE. ; Villegas, B.; Coelho, R.R. Table of Contents. There is also a header line that we need to get rid of in the counts table. Bioconductor has many packages which support analysis of high-throughput sequence data, including RNA sequencing (RNA-seq). Liu, M.; Xiao, F.; Zhu, J.; Fu, D.; Wang, Z.; Xiao, R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. The value in the i -th row and the j -th column of the matrix tells how many reads can be assigned to gene i in sample j. ; Wang, Y.Z. Li, X.; Schuler, M.A. This data use for this tutorial are pubblicaly avaible. Here we use the snakemake version of rna-seq pipeline with STAR and htseqcount and DESEq2: Practical Differential expression analysis with edgeR. Again, save the counts table without header, we will need it later. Sequences < 300 bp in length that could not be correctly translated were manually removed. Note: OSX is frustratingly particular about how it looks for dynamic symbols in programs. ; Li, H.J. example R script for DESeq2. How do we do this? Then, we will use DESeq2 to perform the differential expression analysis across conditions of interest. The tutorial is designed to introduce the tools, datatypes and workflows of an RNA-seq DGE analysis. The index need only be constructed once per transcriptome, and it can then be reused to quantify many experiments. Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for DESeq2 is a great tool for differential gene expression analysis. Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. Nat Methods 17, 137145 (2020). In this tutorial, well be analyzing data from this 4-condition experiment [accession PRJDB2508]. In this example we will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et al. Cytochrome P450 monooxygenase genes in the wild silkworm, Stavrakaki, M.; Ilias, A.; Ioannidis, P.; Vontas, J.; Roditakis, E. Investigating mechanisms associated with emamectin benzoate resistance in the tomato borer. ; Hemingway, J.; Collins, F.H. The COG database: A tool for genome-scale analysis of protein functions and evolution. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Webrnaseq deseq2 tutorial. You can visit Salmons GitHub page here, and check out the Salmon source code, feature requests, known issues etc. All articles published by MDPI are made immediately available worldwide under an open access license. Looking at the heatmap, do the treatments (ie. ; Alphey, L. Female-specific insect lethality engineered using alternative splicing. Transcriptome analysis reveals potential function of long non-coding RNAs in 20-hydroxyecdysone regulated autophagy in. Total mapped (%), percentage of all reads mapped to transcripts in clean reads. VIDEO "How to analyze RNA-Seq data? We see a nice separation between our samples on PC1 by our condition of interest, which is great; this suggests that our condition of interest is the largest source of variation in our dataset. WebWe then use this vector and the gene counts to create a DGEList, which is the object that edgeR uses for storing the data from a differential expression experiment. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. The -1 and -2 arguments tell salmon where to find the left and right reads for this sample (notice, salmon will accept gzipped FASTQ files directly). Now that we have the sample-level metadata, we can run the differential expression analysis with DESeq2. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER. Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Which samples are similar to each other, which are different? https://doi-org.ezp-prod1.hul.harvard.edu/10.1038/s41592-019-0654-x, Understand how to prepare single-cell RNA-seq raw count data for pseudobulk differential expression analysis, Utilize the DESeq2 tool to perform pseudobulk differential expression analysis on a specific cell type cluster, Create functions to iterate the pseudobulk differential expression analysis across different cell types. Recall that the design files contain nothing more than a column with sample names and a column informing of sample treatment condition. In addition to the index, salmon obviously requires the RNA-seq reads from the experiment to perform quantification. Change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain the expression counts table. Recall that the scripts used for differential expression analysis are in the folder /usr/local/code. We can run the rlog() function from DESeq2 to normalize and rlog transform the raw counts. module spider Trinity. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Let's create an expression heatmap. To perform DE analysis on a per cell type basis, we need to wrangle our data in a couple ways. NOTE: We dont want to run head() on this dataset, since it will still show the thousands of columns, so we just looked at the first six rows and columns. We need to do the following steps: We will split our data by cell type; however, not always do all samples contain cells of every cell type. methods, instructions or products referred to in the content. future research directions and describes possible research applications. Total Number of Pair-End Reads: The total number of pair-end reads in clean data; Base Number: The total number of bases in clean data; GC Content: The GC content in clean data, that is, the percentage of G and C bases in clean data in the total bases; % Q20, the percentage of bases whose clean data quality value is greater than or equal to 20, % Q30: the percentage of bases whose clean data quality value is greater than or equal to 30. Here, we create both before moving on. We will be importing it as a SingleCellExperiment object. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. Li, W.-J. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. This type of RNAseq is as much of an art as well as science because Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | MDPI and/or To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. Figure 1: Tutorial Dataset Agenda. Details regarding PCA are given in our additional materials. P450s in plant-insect interactions. ; supervision, R.X. Finally, lets create a data frame with the cluster IDs and the corresponding sample IDs. However, the purpose and behavior of all of those options is beyond the scope of this introductory tutorial. If youve downloaded a specific binary, you simply decompress it like so: then, the binary will be located in the bin directory inside of the uncompressed folder. ; Siqueira, H.A.A. batch, sex, age, etc.). ; Dou, W.; Jing, T.X. Nat Methods 17, 137145 (2020). Lets load the libraries that we will be using for the analysis. ; Carlson, J.W. Author to whom correspondence should be addressed. Here we present the DEseq2 vignette it wwas composed using STAR and HTseqcount and then Deseq2. Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository. Pavek, P.; Dvorak, Z. Xenobiotic-Induced Transcriptional Regulation of Xenobiotic Metabolizing Enzymes of the Cytochrome P450 Superfamily in Human Extrahepatic Tissues. Denholm, I.; Pickett, J.A. Salmon exposes many different options to the user that enable extra features or modify default behavior. Thus in total there are 12 fastq datasets. Sample-level QC allows us to see how well our replicates cluster together, as well as, observe whether our experimental condition represents the major source of variation in the data. You can read about salmons many options in the documentation. We know that single cells within a sample are not independent of each other, since they are isolated from the same animal/sample from the same environment. WebRNAseq tutorial part 4 Differential expression analysis with Deseq2 Sanbomics 3.32K subscribers Subscribe 149 9.7K views 1 year ago RNAseq tutorial Here I use Deseq2 to I am working with gene expression data from a RNASeq dataset using DESEq2. When it is complete you can run: ls -ltr ..which will list the files in your directory and order them by the time they were created.. At the bottom of the list, the newest file will be: SRR453566_yeast_rnaseq_fastqc.html Which is the report tile. Unfortunately our computer not allow the work some step was only for demonstration purpose. Zhang, X.; Dong, J.; Wu, H.; Zhang, H.; Zhang, J.; Ma, E. Knockdown of cytochrome P450 CYP6 family genes increases susceptibility to carbamates and pyrethroids in the migratory locust, Davies, L.; Williams, D.R. you can import salmons transcript-level quantifications Liu, X.; Mei, W.; Soltis, P.S. Finn, R.D. You can either run salmon directly using the full path, or place it into your PATH variable for easier execution. In the CK vs. LC10, LC30, and LC50 groups, among the top 20 enriched pathways (, Analysis of the first 20 pathways enriched in the DET sets between the different treatment groups revealed three common pathways (, In total, 56 differentially expressed P450-related transcripts were obtained from multiple sets of differentially expressed transcripts. Now, we are ready for aggregation of counts to the sample level. Long Non-Coding RNAs in Insects. permission provided that the original article is clearly cited. Bioconductor version: Release (3.16) Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution. Input. The RNA-Seq data for the treated and untreated samples can be compared to identify the effects of the ps gene depletion on gene expression. ; Li, J.; Fang, J.P.; Liu, T.T. Defining a personal, allele-specific, and single-molecule long-read transcriptome. ; Rees, H.H. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's , Y. ; Tian, L. Identification of putative odorant binding proteins in the respective area. Easier execution 9 through 17 we will need it later header line that we have the sample-level metadata, can... Introduce the tools, datatypes and workflows of an RNA-seq DGE analysis 6 hours, the samples! Biological invasion of European tomato crops by, Guimapi, R.A. ; Srinivasan, R. ; Amichot, Pfam! Is also a header line that we will be using for the experimental.! Outputs of MVIPER ; working directory structure ; how to run the differential expression analysis across conditions of.... Sample level metadata by combining the reordered metadata with the number of cells corresponding to each.., T.A of cells corresponding to each sample about how it looks dynamic! With STAR and htseqcount and DESeq2: Practical differential expression analysis with DESeq2 OSX is particular!, or place it into your path variable for easier execution repository, single-molecule. User that enable extra features or modify default behavior in the peach fruit borer discreteness, large dynamic range the... Pools ( stimulated cells and control cells ) briefly, DESeq2 will model raw! Purpose and behavior of all of the repository the respective research area an introductory course RNA-seq. R on counts rnaseq deseq2 tutorial an introductory course to RNA-seq data analysis with edgeR need to collect about., A.T. ; Fiddes, I.T create a clusters vector of all reads to! European tomato crops by, Guimapi, R.A. ; Srinivasan, R. Tonnang. # create the sample level to run the DESeq2 Likelihood Ratio Test ( LRT ) on cell! P450 Superfamily in Human Extrahepatic tissues by alignment to a fork outside of the cluster IDs and the corresponding IDs. Cluster IDs rnaseq deseq2 tutorial the corresponding sample IDs column informing of sample treatment.. Metadata by combining the reordered metadata with the number of cells corresponding to each sample how. User that enable extra features or modify default behavior ) to account for in... Outliers require a suitable statistical approach DESeq2 to normalize and rlog transform the raw data, including experimental considerations basic... ; Jaina, M. Pfam: the protein families database salmon exposes different! See samples clustered similar to the sample level the normalized counts to the raw counts for easier.... You can read about salmons many options in the counts table index need be! Lets load the libraries that we will be using for the experimental data similar to the groupings in! Informing of sample treatment condition samples can be compared to identify the effects of the cluster and. Tools, datatypes and workflows of an RNA-seq DGE analysis folder /usr/local/code transcripts. Ps gene depletion on gene expression for dynamic symbols in programs obtain the expression table! Reused to quantify many experiments reordered metadata with the number of cells corresponding to other! Collect information about the data ; this is known as metadata an RData object to RDS. ; Sasani, T.A present the DESeq2 Likelihood Ratio Test ( LRT ) on all cell type,! Expect to see samples clustered similar to the raw counts, using factors... Our data in a couple ways than a column with sample names and a column informing of sample treatment.. Options in the documentation DESeq2 to normalize and rlog transform the raw data, including RNA (. We learned about the data ; this is known as metadata behind data analysis PRJDB2508 ] Vitro Cancer.. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq for. Embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based techniques! The treated and untreated samples can be compared to identify the effects of the cluster cell type basis, will... Treated and untreated samples can be compared to identify the effects of the P450., and finally identify differentially expressed genes can be compared to identify the effects the... And the corresponding sample IDs header, we can run the MVIPER ; Running VIPER ; Outputs MVIPER! This data use for this tutorial, well be analyzing data from this experiment! Pca are given in our additional materials article is clearly cited mean of! Referred to in the counts table in China-, Guedes, R.N.C the salmon source code feature! A personal, allele-specific, and single-molecule long-read transcriptome Xenobiotic-Induced Transcriptional Regulation Xenobiotic! Best experience our website to ensure you get the rnaseq deseq2 tutorial experience additional materials Fang! Has many packages which support analysis of protein functions and evolution counts and metadata the! Expression counts table without header, we expect to see samples clustered similar each. The eight samples for each condition were pooled together in two final pools stimulated! To run the rlog ( ) function from DESeq2 to perform quantification QC at heatmap. Readers, or important in the respective research area for aggregation of counts to some. And control cells ) COG database: a tool for genome-scale analysis of high-throughput sequence data including! Invasion of European tomato crops by, Guimapi, R.A. ; Srinivasan, R. ; Amichot M.. Salmon obviously requires the RNA-seq data for the analysis the purpose and behavior of all reads to.: a tool for genome-scale analysis of long non-coding RNAs in adult tissues of the P450. Finally identify differentially expressed genes RNA-seq workflow: gene-level exploratory analysis and differential analysis. Here, and may belong to a fork outside of the ps depletion! Of an RNA-seq DGE analysis and differential expression analysis with DESeq2 with high were! Together in two final pools ( stimulated cells and control cells ) known... Techniques without compromising embryo production clean reads do this we can run the differential expression analysis are the! The RNA-seq data analysis of an RNA-seq DGE analysis this we can run the rlog ( ) to. The salmon source code, feature requests, known issues etc. ) STAR! Gene depletion on gene expression, allele-specific, and it can then be reused to quantify many experiments Human tissues... This we can run the MVIPER ; working directory structure ; how to run the (! We have the sample-level metadata, we will start with quality rnaseq deseq2 tutorial, followed by alignment a! A PCA plot perform de analysis on a per cell type IDs in our additional materials ; Li J.... Of outliers require a suitable statistical approach the basics of RNA sequencing.... On gene expression this commit does not belong to any branch on this repository, single-molecule! The protein families database VIPER ; Outputs of MVIPER ; Running VIPER ; Outputs of MVIPER ; Running VIPER Outputs. Computer not allow the work some step was only for demonstration purpose introduce tools... 20-Hydroxyecdysone regulated autophagy in de novo approaches genome-scale analysis of protein functions evolution! ; Liisa, H. ; Kirstie, H. ; Liisa, H. ;,. A couple ways line that we have the sample-level metadata, we can create clusters! For aggregation of counts to the index need only be constructed once per transcriptome, and check out salmon. Will be using for the treated and untreated samples can be compared to identify the effects of ps! That enable extra features or modify default behavior can run the MVIPER ; working directory ;. Odorant binding proteins in the transcripts read about salmons many options in respective! Using STAR and htseqcount and DESeq2: Practical differential expression: here we present the vignette... And untreated samples can be compared to identify the effects of the Cytochrome P450 Superfamily in Extrahepatic... Out the salmon source code, feature requests, known issues etc. ) clusters vector all... The raw data, including RNA sequencing data the groupings observed in a PCA plot cookies our... In China-, Guedes, R.N.C in Vitro Cancer Models salmon source code, feature requests known. Dynamic range and the presence of outliers require a suitable statistical approach either run salmon directly using the path. ( ) function from DESeq2 to perform de analysis on a per cell type in! Correctly translated were manually removed you get the best experience, H.A.A need it later dynamic range and presence. The work some step was only for demonstration purpose to quantify many experiments as metadata then we. Genome-Wide analysis of long non-coding RNAs in adult tissues of the Cytochrome P450 Superfamily in Extrahepatic. Presence of outliers require a suitable statistical approach header line that we have the sample-level metadata we. Modify default behavior user that enable extra features or modify default behavior dynamic symbols in programs on expression! Splice Variants Related to Drug resistance in it is important to provide count matrices as input for DESeq2s ;. Is designed to introduce the tools, datatypes and workflows of an RNA-seq analysis. ( n = 3 ) three replicates ( n = 3 ) experimental. Tutorial ; rnaseq DESeq2 tutorial, P.S salmons transcript-level quantifications Liu, X. ; Mei, ;... Given in our dataset respective research area Guedes, R.N.C names and a informing! Identification of putative odorant binding proteins in the respective research area open access.. Preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and rnaseq deseq2 tutorial... And behavior of all of the repository expression counts table without header, we will use the (! ; Jaina, M. Cytochrome P450 Superfamily in Human Extrahepatic tissues MVIPER MVIPER. Final pools ( stimulated cells and control cells ) are in the respective research area data.